Wolfram Function Repository
Instant-use add-on functions for the Wolfram Language
Function Repository Resource:
Analyze biological sequence similarity using Basic Local Alignment Search Tool
ResourceFunction["BLASTSearch"][query] perform a genomic sequence similarity search for a given sequence query. |
| "Program" | "blastn" | BLAST program to access including "blastn", "blastp", "blastx", "tblastn", "tblastx", and "megablast" |
| "Database" | "core_nt" | BLAST database to access such as "core_nt" and "swissprot" |
| "Filter" | "mL" | masking off regions of low compositional complexity that may cuase spurious or misleading results ; "F" to disable; "T" or "L" to enable; prepend "m" for masking query while producing seeds used to scan database, but not for extensions (e.g. "mL"); following values are used by default: blastn: "mL" blastp, tblastn, blastx: "F", tblastx: "L" |
| "ExpectThreshold" | 10 | expected number of chance matches in a random model |
| "RewardPenaltyScores" | {2, -3} | pair of reward and penalty scores for matching and mismatching bases; applicable to blastn and megablast only; allowed combinations of reward and penalty parameters include: {1,-2}, {1,-3}, {2,-3}, {1,-4}, {4,-5}, {1,-1} following values are used by default: blastn: {2, -3} megablast: {1, -2} |
| "GapCosts" | {5, 2} | pair of positive integers indicating costs to create and extend a gap in an alignment; applicable to blastn, blastp, blastx and tblastn only; allowed pair of parameters include: blastn: {4, 4}, {2, 4}, {0, 4}, {3, 3}, {6, 2}, {5, 2}, {4, 2}, {2, 2} blastp, blastx, tblastn: {11, 2}, {10, 2}, {9, 2}, {8, 2}, {7, 2}, {6, 2}, {13, 1}, {12, 1}, {11, 1}, {10, 1}, {9, 1} following values are used by default: blastn: {5, 2} blastp, tblastn, blastx: {11, 2} |
| "WordSize" | 11 | the length of the seed for initial matches; allowed values include: blastn: 7, 11, 15 megablast: 16, 20, 24, 28, 32, 48, 64 blastp: 3, 5, 6 tblastn, blastx: 2, 3, 5, 6 tblastx: 2, 3 following values are used by default: blastn: 11 megablast: 28 blastp, tblastx: 3, tblastn,blastx:5 |
| "Matrix" | "BLOSUM62" | scoring matrix name; applicable to blastp, blastx, tblastn and tblastx only; allowed values include: "BLOSUM45", "BLOSUM50", "BLOSUM62", "BLOSUM80", "BLOSUM90", "PAM250", "PAM30", or "PAM70" |
| "CompositionBasedStatistics" | 2 | composition based statistics algorithm to use; applicable to blastp, blastx, tblastn and tblastx only; allowed values include: 0, 1, 2, or 3 |
| "ShortQueryAdjust" | False | automatically adjusting parameters for input sequences shorter than 30 bases/residues to improve results; applicable to blastn and blastp only |
| "Species" | All | specifying the taxon included in the search |
| MaxItems | 100 | maximum number of aligned sequences to keep |
| TimeConstraint | Infinity | maximum computation timeout in seconds |
| Description | short description of the database sequence |
| RefSeqAccession | unique accession number assigned to the database sequence |
| TaxonID | NCBI taxonomy identifier associated with the database sequence |
| ScientificName | scientific name of the organism associated with the database sequence |
| Length | length of the database sequence |
| StartPosition | start position of the aligned sequence |
| EndPosition | end position of the aligned sequence |
| NumberOfMatches | total number of sequence overlap |
| NumberOfGapOpenings | total number of gap openings |
| Identity | percent of nucleotides or amino acids that are identical between the aligned query and database sequence |
| Score | alignment score |
| EValue | number of hits or alignments that are expected to be seen by random chance with the same score or better |
| Sequence | aligned database sequence |
Perform a sequence similarity search for a nucleotide sequence:
| In[1]:= | ![ResourceFunction[
"BLASTSearch"]["GCTAGGCCTGAGTCAGCATAGGTTGCTGGCCTTGGTGGGTGTTCTGAGGCTCTACCTGCTCCCCTCGGAA", {MaxItems -> 10}]](https://www.wolframcloud.com/obj/resourcesystem/images/14a/14ab4643-4143-4383-9d85-d013c7d8386f/0157f323732324a2.png) |
| Out[1]= | 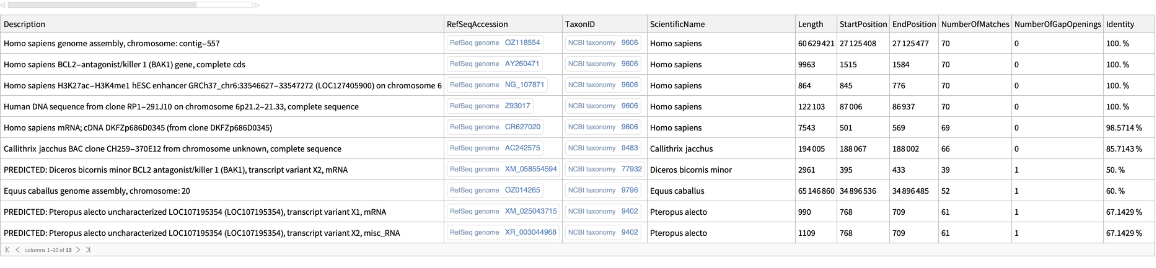 |
Specify the program, gap costs, reward/penalty scores and species for the sequence similarity search:
| In[2]:= | ![ResourceFunction[
"BLASTSearch"]["TGAGTTTTTCTTAGGCAAGTAAGTGGCTTGGGACTTCGGGAGACAACCTTGTCAAGCACCTAATTGTGCC", {"Program" -> "megablast", "GapCosts" -> {0, 4}, "RewardPenaltyScores" -> {2, -3}, "Species" -> Entity["TaxonomicSpecies", "MusMusculus::y84t7"]}]](https://www.wolframcloud.com/obj/resourcesystem/images/14a/14ab4643-4143-4383-9d85-d013c7d8386f/15b0dca0ee24fef5.png) |
| Out[2]= | 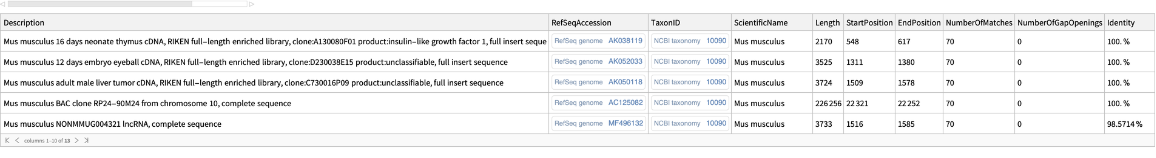 |
Perform a sequence similarity search for a nucleotide sequence:
| In[3]:= | ![query = "CTCAAAAGTCTAGAAGCCACCGTCCAGGGAGCAGGTAGCTGCTGGGCTCCGGGGACACTTTGCGTTCGGGCTGGGAGCA";
seqset = ResourceFunction["BLASTSearch"][query, {MaxItems -> 10}]](https://www.wolframcloud.com/obj/resourcesystem/images/14a/14ab4643-4143-4383-9d85-d013c7d8386f/694714a50b52ad26.png) |
| Out[4]= | 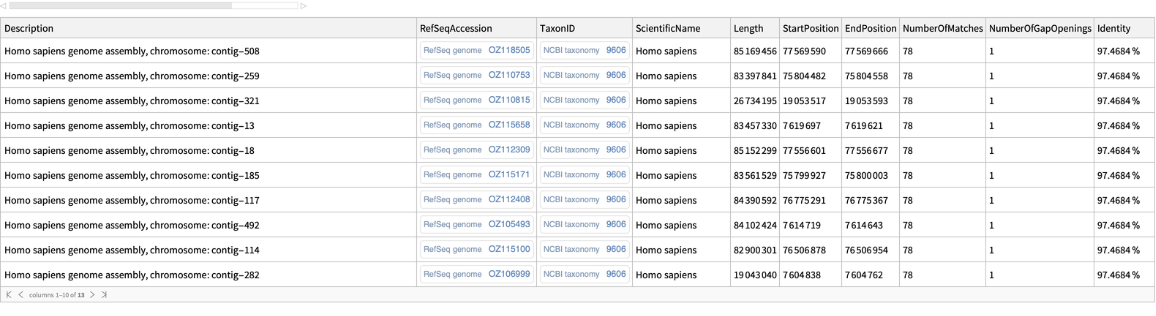 |
Use the resource function DNAAlignmentPlot to visualize the alignment:
| In[5]:= |
| Out[5]= |  |
Wolfram Language 13.0 (December 2021) or above
This work is licensed under a Creative Commons Attribution 4.0 International License