Wolfram Function Repository
Instant-use add-on functions for the Wolfram Language
Function Repository Resource:
Retrieve information on reference SNPs from the NCBI database
ResourceFunction["NCBIGenomicSNPData"][snp, "VariantDetails"] gives the dataset of variant information for a specified snp. | |
ResourceFunction["NCBIGenomicSNPData"][snp, "FrequencyData"] gives the dataset of allele frequencies for a specified snp. | |
ResourceFunction["NCBIGenomicSNPData"][snp, "ClinicalSignificance"] gives the dataset of associated diseases for a specified snp. |
| "Gene" | associated ID |
| "GeneSymbol" | associated gene symbol |
| "Orientation" | orientation of the genomic sequence |
| "NucleotideSeqAccession" | NCBI nucleotide sequence accession ID |
| "NucleotidePosition" | position of the allele on the nucleotide sequence |
| "DeletedSequence" | sequence of deleted nucleotides or the codon |
| "InsertedSequence" | sequence of inserted nucleotides or the codon |
| "NucleotideVarSequenceOntologyAccession" | accession ID of the Sequence Ontology (SO) concept describing the nucleotide sequence variation |
| "NucleotideVarSequenceOntologyTerm" | name of the Sequence Ontology (SO) concept describing the nucleotide sequence variation |
| "HGVS" | Human Genome Variation Society (HGVS) notation |
| "ProteinSeqAccession" | NCBI protein sequence accession ID |
| "ProteinPosition" | position of the amino acid change on the protein sequence |
| "DeletedAminoAcid" | letter of the deleted amino acid |
| "InsertedAminoAcid" | letter of the inserted amino acid |
| "ProteinVarSequenceOntologyAccession" | accession ID of the Sequence Ontology (SO) concept describing the protein sequence variation |
| "ProteinVarSequenceOntologyTerm" | name of the Sequence Ontology (SO) concept describing the protein sequence variation |
| "StudyName" | name of the study |
| "RefSeqAccession" | NCBI refrence sequence accession ID |
| "Position" | position of the allele on the reference sequence |
| "RefAllele" | reference allele |
| "AltAllele" | alternate allele |
| "RefAlleleFrequency" | reported reference allele frequency |
| "AltAlleleFrequency" | reported alternate allele frequency |
| "TotalCount" | total sample size |
For SNP RS429358, which is a genetic variation found in the APOE gene associated with a risk of Alzheimer's disease, list variant details:
| In[1]:= |
| Out[1]= | 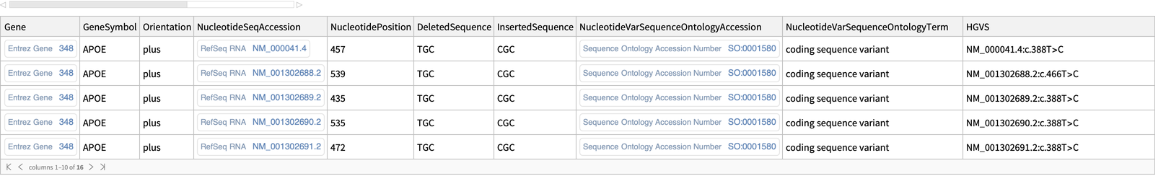 |
Retrieve clinical significance information for a given SNP:
| In[2]:= |
| Out[2]= | 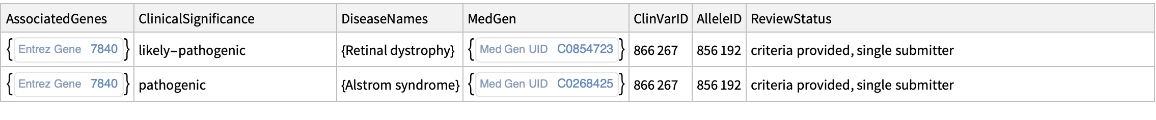 |
Retrieve allele frequency data for a given SNP entity:
| In[3]:= |
| Out[3]= | 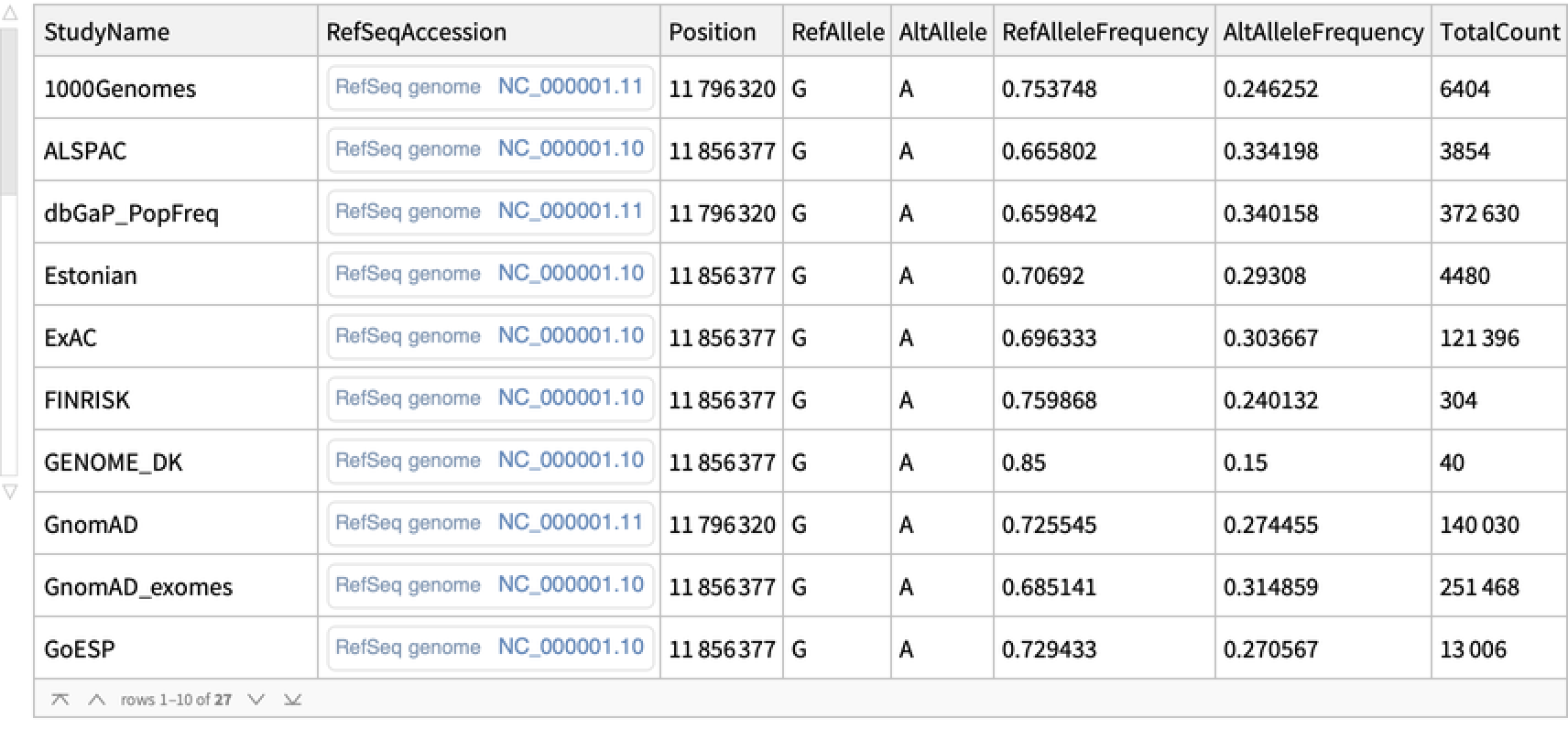 |
Compare the reference and the alternate sequences associated with a given SNP:
| In[4]:= |
| Out[4]= |
Use the ImportFASTA ResourceFunction to retrieve the reference sequence:
| In[5]:= |
| In[6]:= |
| Out[6]= | 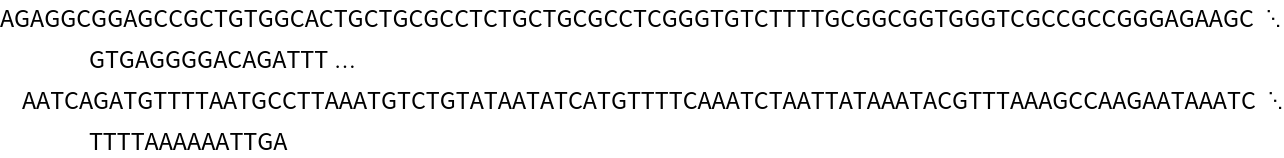 |
Compute the alternate sequence using "NucleotidePosition", "DeletedSequence" and "InsertedSequence" information:
| In[7]:= | ![alt = StringReplacePart[ref, brca[1, "InsertedSequence"], {brca[1, "NucleotidePosition"], brca[1, "NucleotidePosition"] + StringLength[brca[1, "DeletedSequence"]] - 1}];](https://www.wolframcloud.com/obj/resourcesystem/images/6f2/6f2d5756-cc3b-42f3-932f-bc6d163c3291/1-0-0/2787a22692a4bf70.png) |
Use the DNAAlignmentPlot function to visualize the allele position:
| In[8]:= |
| Out[8]= |  |
Next, explore how this change impacts the part of translated peptide sequences. Apply the BioSequenceTranslate function to the reference sequence to retrieve the sequence of amino acids:
| In[9]:= | ![refPep = BioSequenceTranslate[
BioSequence[
StringTake[
ref, {brca[1, "NucleotidePosition"], brca[1, "NucleotidePosition"] + 14}]]]](https://www.wolframcloud.com/obj/resourcesystem/images/6f2/6f2d5756-cc3b-42f3-932f-bc6d163c3291/1-0-0/63b8ec27ad3971ca.png) |
| Out[9]= |
Notice that the reading frame is shifted for the alternate peptide sequence and the stop codon is inserted four amino acids downstream:
| In[10]:= | ![altPep = BioSequenceTranslate[
BioSequence[
StringTake[
alt, {brca[1, "NucleotidePosition"], brca[1, "NucleotidePosition"] + 14}]]]](https://www.wolframcloud.com/obj/resourcesystem/images/6f2/6f2d5756-cc3b-42f3-932f-bc6d163c3291/1-0-0/6b3c8e77e45f9562.png) |
| Out[10]= |
Compare the molecule plots:
| In[11]:= |
| Out[11]= | 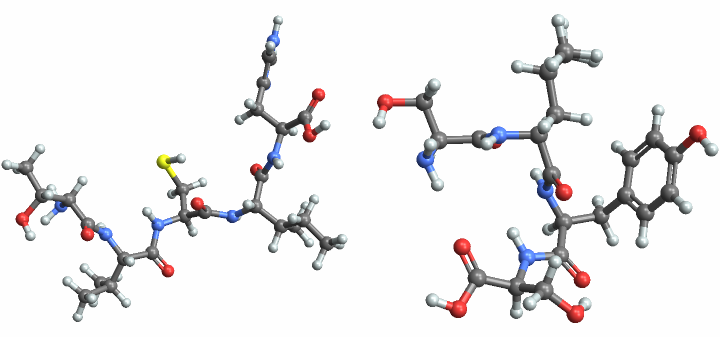 |
Wolfram Language 13.0 (December 2021) or above
This work is licensed under a Creative Commons Attribution 4.0 International License