Wolfram Function Repository
Instant-use add-on functions for the Wolfram Language
Function Repository Resource:
Import files in the VCF format, a bioinformatics standard for storing gene sequence variations
ResourceFunction["ImportVCF"][file] imports file as a VCF file. |
Import a VCF sample file from GitHub:
| In[1]:= |
| Out[1]= |  |
Import another sample file:
| In[2]:= |
| Out[2]= | 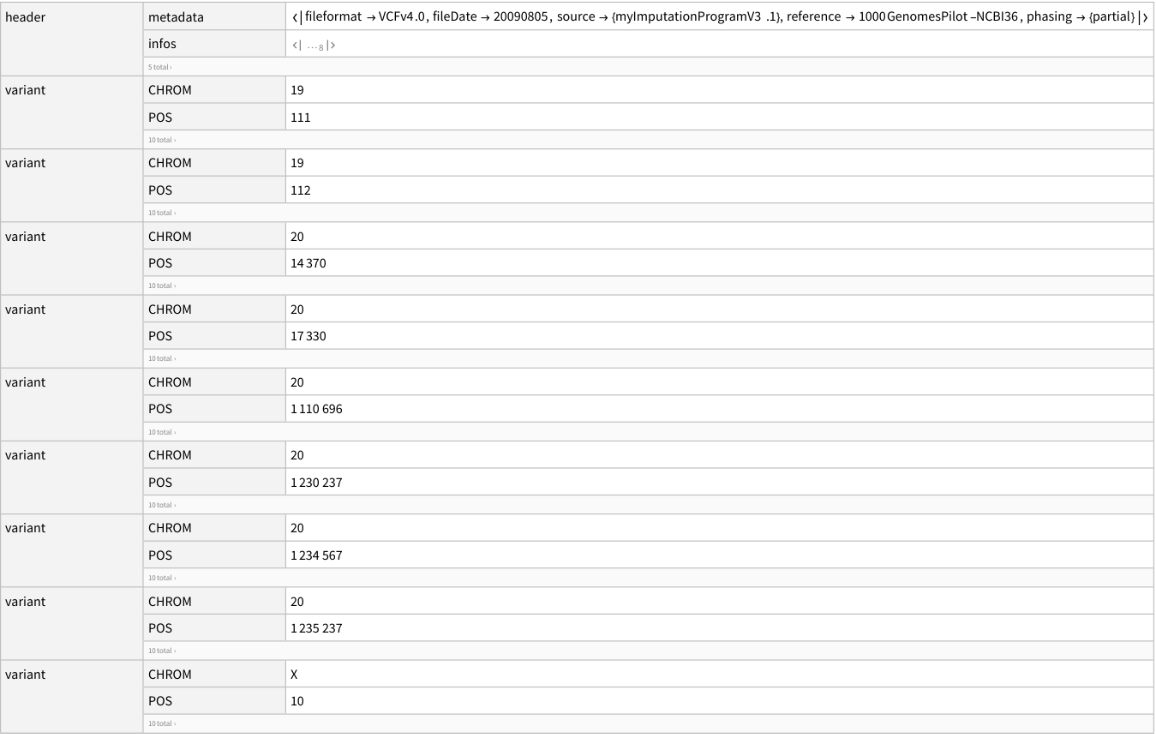 |
Obtain the human variation set for GRCh38:
| In[3]:= |
Extract the compressed archive file (contains one file):
| In[4]:= |
Import the VCF file (this takes a few minutes due to the large file size):
| In[5]:= |
Examine the first 50 rows of data:
| In[6]:= |
| Out[6]= | 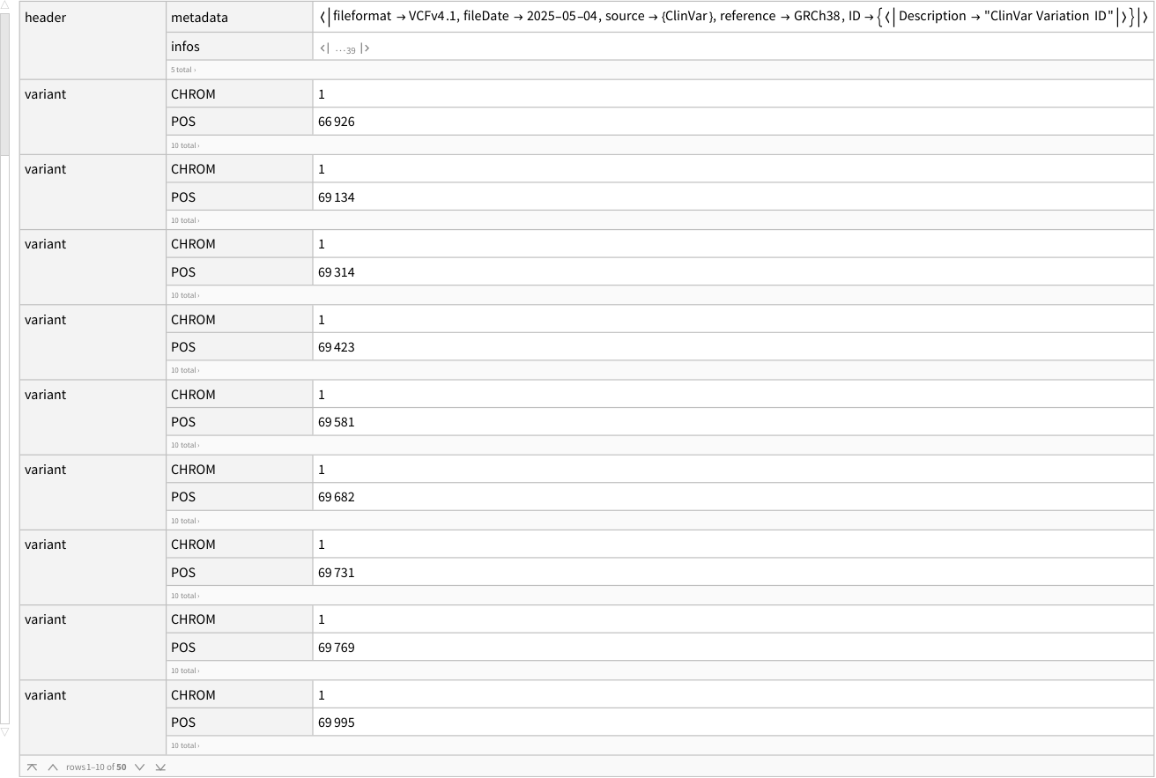 |
Wolfram Language 14.0 (January 2024) or above
This work is licensed under a Creative Commons Attribution 4.0 International License