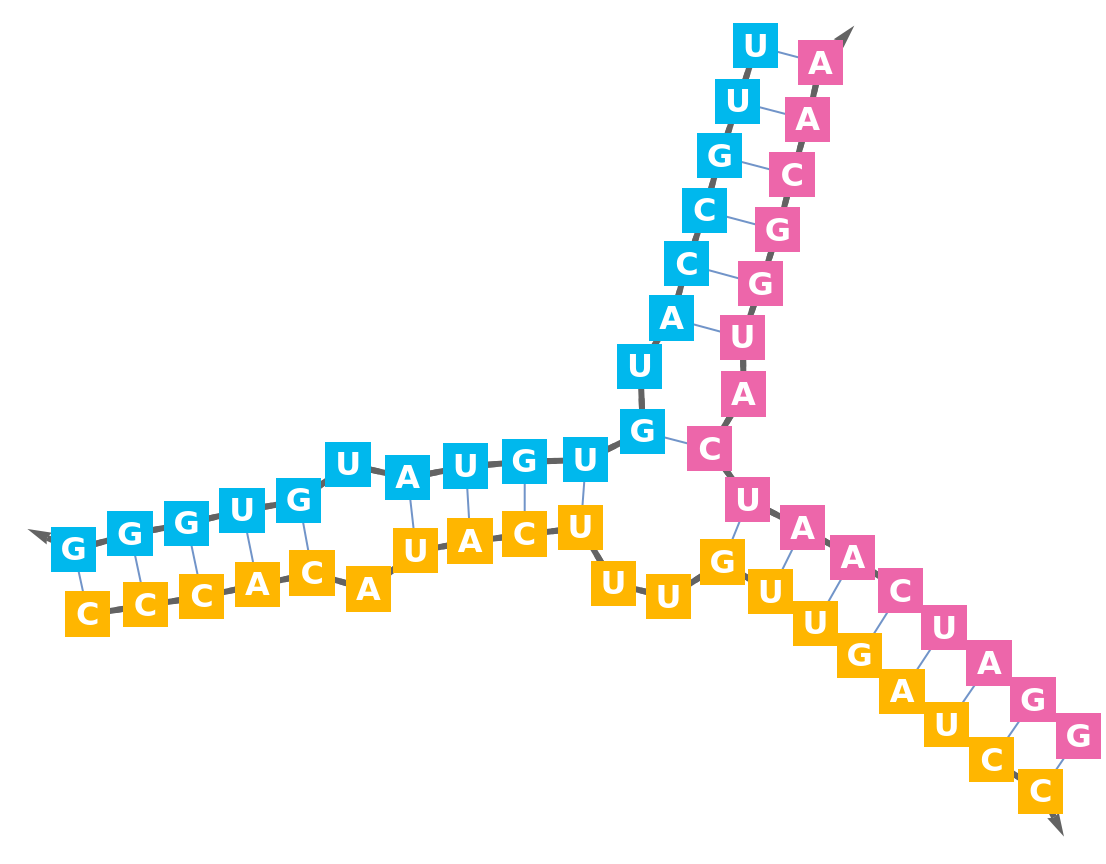Basic Examples (3)
Import the structure for the yeast phenylalanine tRNA:
Visualize the molecule:
Find the base-pairs:
Scope (2)
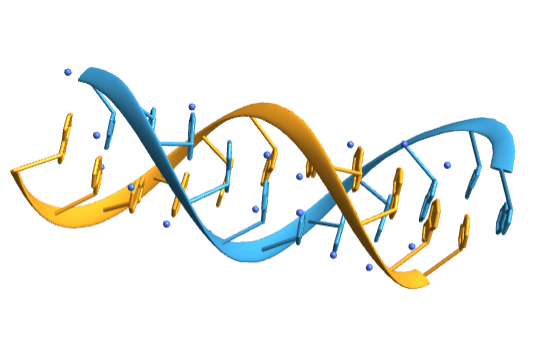
Import a duplex RNA:
Base-pairs between chains are found when more than one nucleic acid polymer is present in the BioMolecule:
Find the base-pairs:
Import a short RNA that has four models:
Base-pairs can be found for different models in a BioMolecule:
Find the base-pairs for all the models:
Options (12)
MaxOriginDistance (3)
Import an RNA biomolecule:
Compute the base-pairs with the default criteria:
Use a severely constrained "MaxOriginDistance" criterion:
MaxVerticalSeparation (3)
Import an RNA biomolecule:
Compute the base-pairs with the default criteria:
Use a severely constrained "MaxVerticalSeparation" criterion:
MaxBaseNormalsAngle (3)
Import an RNA biomolecule:
Compute the base-pairs with the default criteria:
Use a severely constrained "MaxBaseNormalsAngle" criterion:
MinHydrogenBondCount (3)
Import an RNA biomolecule:
Compute the base-pairs with the default criteria:
Use a relaxed "MinHydrogenBondCount" criterion:
Applications (6)
Nucleic acid base-pairs can be used to construct the sequence bonds of a BioSequence. Import a short RNA strand that forms a simple loop:
The sequence bonds are not present in the BioMolecule:
Find the positions of the base-pairs:
Construct the BioSequence with "Multihydrogen" bonds:
Display the secondary structure:
Display the biomolecule with the base-pairs colored:
Possible Issues (4)
Atom labels in biomolecules are generally standardized, but exceptions are known, so the function may occasionally fail to find the atoms of a nucleotide base and therefore be unable to compute the base-pair parameters. When that occurs, the residues are removed from further consideration.
Import the structure of 3B5A, a minimally hinged hairpin ribozyme:
Display the residue abbreviations for the RNA chains:
Note the nonstandard residue A2m in chain A and S9l in chain B. Attempt to compute the base pairs:
Count the intra- (A-A, B-B, C-C) and inter-strand (A-B, A-C, B-C) base pairs:
Neat Examples (2)
Base-pairing in RNA can be quite intricate, and many topologies are known.
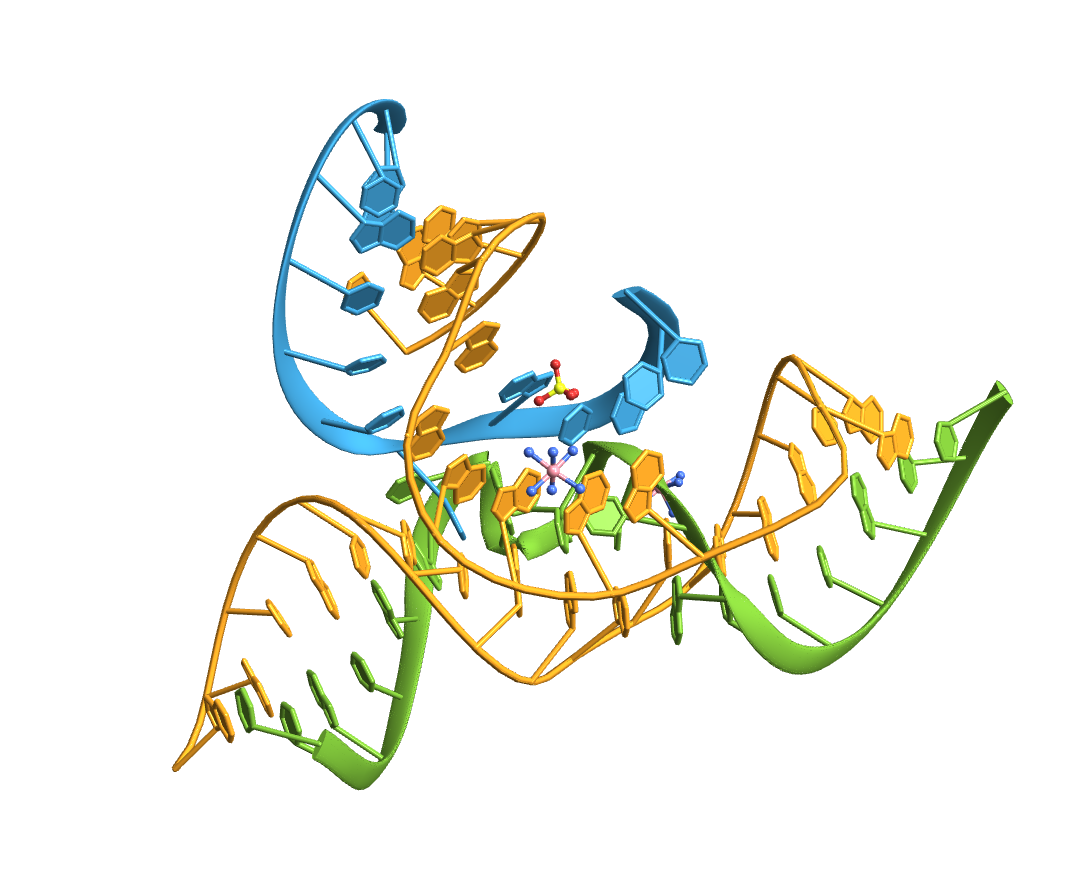
Import the structure of an RNA "kissing" hairpin complex of the HIV TAR hairpin loop and its complement:
Compute the base-pairs:
Count the intra- (A-A, B-B) and inter-strand (A-B) base pairs:
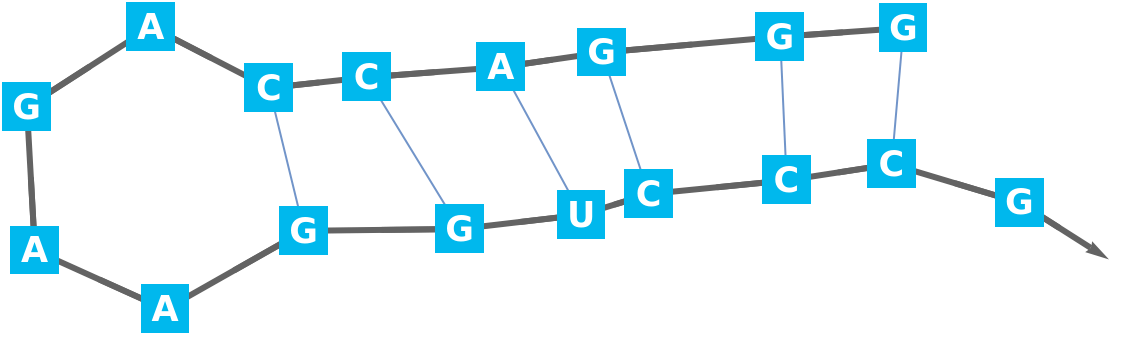
Construct the BioSequence:
Display the secondary structure:
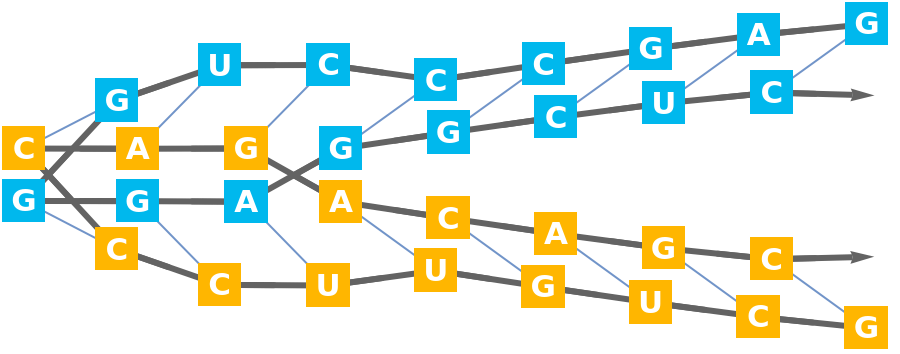
Import an RNA structure with a three way junction (3WJ):
Compute the base-pairs:
Count the intra- (A-A, B-B, C-C) and inter-strand (A-B, A-C, B-C) base pairs:
Construct the BioSequence:
Display the secondary structure:



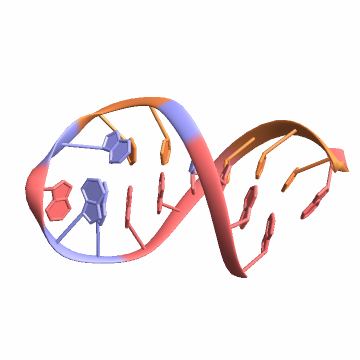

![biomol = BioMolecule[ExternalIdentifier["PDBStructureID", "1ATV"]];
BioMoleculePlot3D[biomol, ImageSize -> Small, ColorRules -> "Residues"]](https://www.wolframcloud.com/obj/resourcesystem/images/d1a/d1aeecf0-584e-4f55-b06d-0dc8f1579d79/7fde030c184726f6.png)
![bioseq = BioSequence[biomol["BioSequences"]["A"]["SequenceType"], biomol["BioSequences"]["A"]["SequenceString"], Bond[#, "MultiHydrogen"] & /@ basePairs[[All, All, 3]]]](https://www.wolframcloud.com/obj/resourcesystem/images/d1a/d1aeecf0-584e-4f55-b06d-0dc8f1579d79/103babd708eb2b6a.png)
![BioMoleculePlot3D[biomol, ColorRules -> Append[MapThread[
Splice@Thread[#1 -> #2] &, {basePairs[[All, All, 2 ;; 3]], {StandardRed, StandardBlue, StandardGreen, StandardCyan, StandardYellow, StandardMagenta}}], _ -> StandardGray]]](https://www.wolframcloud.com/obj/resourcesystem/images/d1a/d1aeecf0-584e-4f55-b06d-0dc8f1579d79/35c60cef9f797897.png)
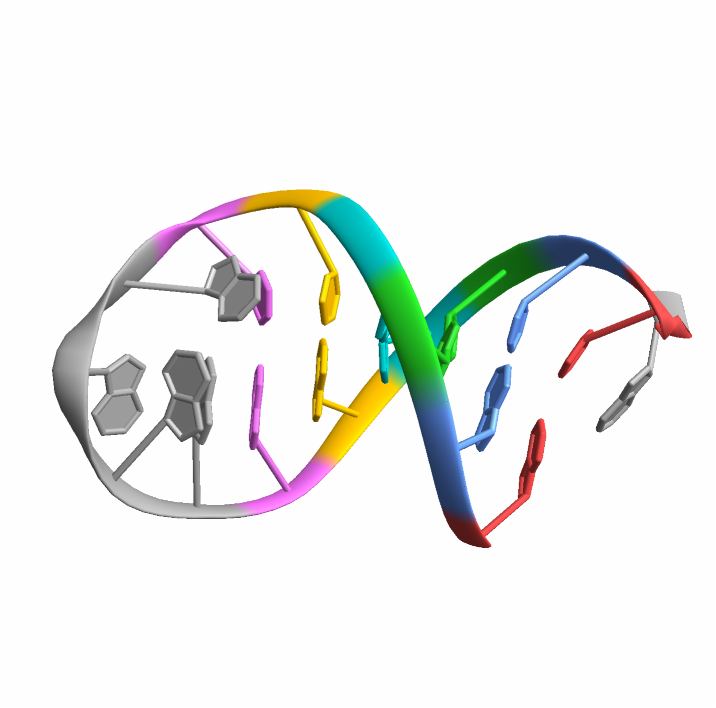




![bioseq = BioSequence[
BioSequence["RNA", #] & /@ StringJoin @@@ Values@biomol["ResidueAbbreviations"],
Bond[#, "MultiHydrogen"] & /@ (basePairs[[All, All, {2, 3}]] /. {"A" -> 1, "B" -> 2})
]](https://www.wolframcloud.com/obj/resourcesystem/images/d1a/d1aeecf0-584e-4f55-b06d-0dc8f1579d79/41285de0efc484b4.png)



![bioseq = BioSequence[
BioSequence["RNA", #] & /@ StringJoin @@@ Values@KeySelect[biomol["ResidueAbbreviations"], biomol["ChainTypes"][#] == "RNA" &],
Bond[#, "MultiHydrogen"] & /@ (basePairs[[All, All, {2, 3}]] /. {"A" -> 1, "B" -> 2, "C" -> 3})
]](https://www.wolframcloud.com/obj/resourcesystem/images/d1a/d1aeecf0-584e-4f55-b06d-0dc8f1579d79/53d662220e1e2327.png)