Wolfram Function Repository
Instant-use add-on functions for the Wolfram Language
Function Repository Resource:
Compute the 166-bit MACCS (Molecular ACCess System) key
ResourceFunction["MACCSKeys"][molecule] returns the MACCS key for the Molecule molecule. | |
ResourceFunction["MACCSKeys"][smiles] returns the MACCS key for a molecule specified by the SMILES string smiles. |
MACCSKeys can take either a SMILES string or a Molecule as input. By default, it returns a SparseArray containing the 166 bits:
| In[1]:= |
| Out[1]= |
| In[2]:= |
| Out[2]= |
MACCSKeys is a Listable function:
| In[3]:= | ![ResourceFunction[
"MACCSKeys"][{"CCO", "O" , "[H][C@@]1([C@@H](C2=CC=NC3=CC=C(C=C23)OC)O)C[C@@H]4CC[N@]1C[C@@H]4C=C"}]](https://www.wolframcloud.com/obj/resourcesystem/images/f8b/f8b0f1a9-223b-4eda-9d8c-9e4928b1b029/35e3df53ab6eb3fd.png) |
| Out[3]= | 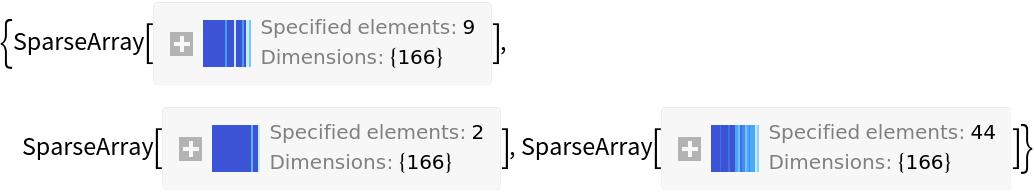 |
Option values include "SparseArray", "OnBits", "MoleculePlot", "Function" and "SMARTS". The default setting of "SparseArray" returns the 166-bit vector:
| In[4]:= |
| Out[4]= |
The "OnBits" setting returns a list of the active (non-zero) bits in the MACCS key. These are 1-indexed (as is conventional in the Wolfram Language):
| In[5]:= |
| Out[5]= |
The "MoleculePlot" setting returns an association whose keys are the active bits and whose values are the MoleculePlots corresponding to the MoleculePattern that was matched for that key. Here we take the first three, for brevity:
| In[6]:= |
| Out[6]= | 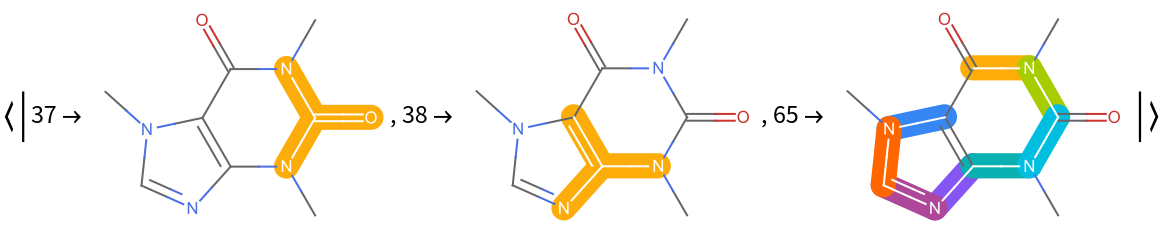 |
The "Function" setting returns an association whose values are pure functions responsible for generating each key:
| In[7]:= |
| Out[7]= |  |
The "SMARTS" setting returns an Association whose values are the SMARTS specification for the pattern. Note that not all MACCS keys can be defined as SMARTS patterns (these return a “?”) and some MACCS keys require finding a certain number of matches above some threshold, so the SMARTS specification alone is not always a complete description of the key:
| In[8]:= |
| Out[8]= |
Compare the structural similarity of six common statin drugs using the JaccardDissimilarity of the MACCS keys (one minus this is equivalent to the Tanimoto similarity):
| In[9]:= | ![statins = <|
"Zocor" -> "CCC(C)(C)C(=O)O[C@H]1C[C@H](C=C2[C@H]1[C@H]([C@H](C=C2)C)CC[C@@H]3C[C@H](CC(=O)O3)O)C", Sequence[
"Pravachol" -> "CC[C@H](C)C(=O)O[C@H]1C[C@@H](C=C2[C@H]1[C@H]([C@H](C=C2)C)CC[C@H](C[C@H](CC(=O)[O-])O)O)O", "Lipitor" -> "CC(C)C1=C(C(=C(N1CC[C@H](C[C@H](CC(=O)[O-])O)O)C2=CC=C(C=C2)F)C3=CC=CC=C3)C(=O)NC4=CC=CC=C4.[Ca+2]", "Lescol" -> "CC(C)N1C2=CC=CC=C2C(=C1/C=C/[C@H](C[C@H](CC(=O)O)O)O)C3=CC=C(C=C3)F", "Crestor" -> "CC(C1=NC(=NC(=C1/C=C/[C@@H](O)C[C@@H](O)CC(=O)[O-])C2=CC=C(C=C2)F)N(S(=O)(=O)C)C)C.CC(C1=NC(=NC(=C1/C=C/[C@@H](O)C[C@@H](O)CC(=O)[O-])C2=CC=C(C=C2)F)N(S(=O)(=O)C)C)C.[Ca+2]", "Altoprev" -> "CC[C@H](C)C(=O)O[C@H]1C[C@H](C=C2[C@H]1[C@H]([C@H](C=C2)C)CC[C@@H]3C[C@H](CC(=O)O3)O)C"]|>; (*define 6 common statin drugs*)
similarity = 1. - DistanceMatrix[
Values@
ResourceFunction["MACCSKeys"]@
statins, (*use the MACCS keys to calculate the (dis)similarity*)
DistanceFunction -> JaccardDissimilarity];
TableForm[similarity, (*display*)
TableHeadings -> {Keys[statins], Keys[statins]}]](https://www.wolframcloud.com/obj/resourcesystem/images/f8b/f8b0f1a9-223b-4eda-9d8c-9e4928b1b029/0777d36ba95098aa.png) |
| Out[527]= | 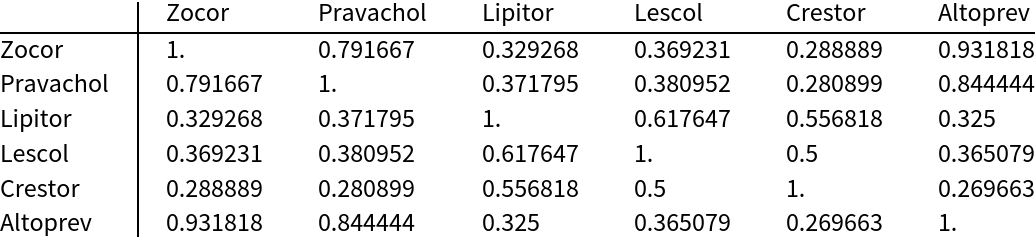 |
Empirically, less than 3% of randomly selected molecules have a MACCS Tanimoto similarity above 0.6. Use this as a threshold to visualize which molecules are similar to one another:
| In[528]:= | ![AdjacencyGraph[
Keys@statins,(*use drug names as vertexes*)
UnitStep[
similarity - IdentityMatrix[6] - 0.6 ], (*only draw edges above threshold*)
VertexLabels -> KeyValueMap[#1 -> Tooltip[#1, Thumbnail@MoleculePlot@Molecule@#2] &,
statins] (*create mouseover graphics with molecule images*)
]](https://www.wolframcloud.com/obj/resourcesystem/images/f8b/f8b0f1a9-223b-4eda-9d8c-9e4928b1b029/075ea892f899ae20.png) |
| Out[528]= | 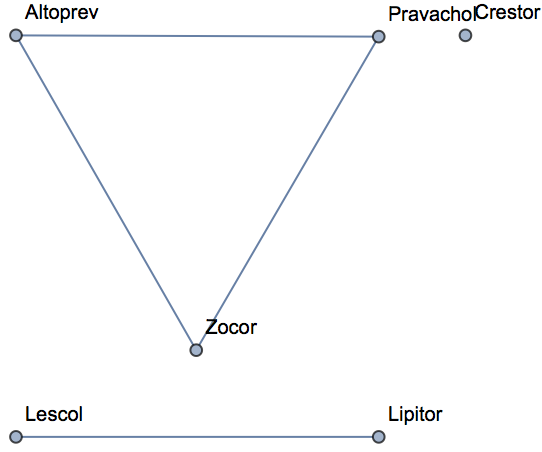 |
Generate random compound IDs and SMILES strings:
| In[529]:= | ![RandomSeed[1841];
cids = RandomInteger[10^8, 200] + 1; (*generate random CompoundIDs*)
smiles = (*Look up SMILES strings for the CIDS*)
ServiceExecute["PubChem", "CompoundProperties", {"CompoundID" -> cids, "Property" -> "IsomericSMILES"}][[All, "IsomericSMILES"]] // Normal;](https://www.wolframcloud.com/obj/resourcesystem/images/f8b/f8b0f1a9-223b-4eda-9d8c-9e4928b1b029/0e2559509e35d87f.png) |
Get their MAACS keys:
| In[530]:= |
The function for Tanimoto similarity:
| In[531]:= |
Use the function to compute the Tanimoto similarites of the random compounds:
| In[532]:= | ![scores = ParallelMap[ (*compute all the pairwise Tanimoto Similarities*)
tanimotoSimilarity[maccs[[#]], maccs[[# + 1 ;; -1]]] &,
Range[199]] // Flatten;](https://www.wolframcloud.com/obj/resourcesystem/images/f8b/f8b0f1a9-223b-4eda-9d8c-9e4928b1b029/6c370452fa6262a5.png) |
Generate a histogram of the similarity scores:
| In[533]:= |
| Out[533]= | 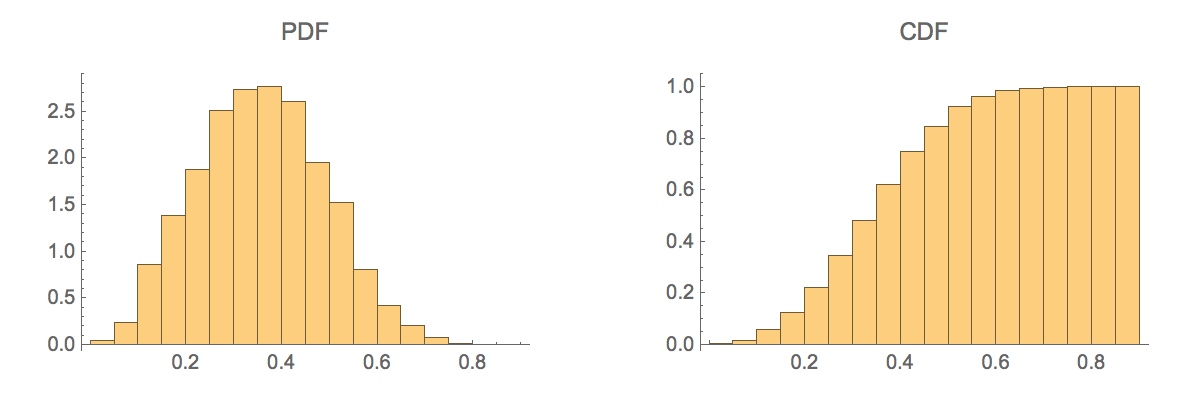 |
The mean Tanimoto similarity score for randomly selected molecules is approximately 0.35:
| In[534]:= |
| Out[534]= |
Only about 3% of randomly chosen molecules will have a Tanimoto similarity score above 0.6:
| In[535]:= |
| Out[535]= |
This work is licensed under a Creative Commons Attribution 4.0 International License