Wolfram Function Repository
Instant-use add-on functions for the Wolfram Language
Function Repository Resource:
Convert a chemical name into a SMILES identifier string
ResourceFunction["ChemicalNameToSMILES"]["name"] returns a SMILES string corresponding to the given chemical name. |
Find the SMILES string from a chemical name:
| In[1]:= |
| Out[1]= |
Plot the molecule in 3D:
| In[2]:= |
| Out[2]= | 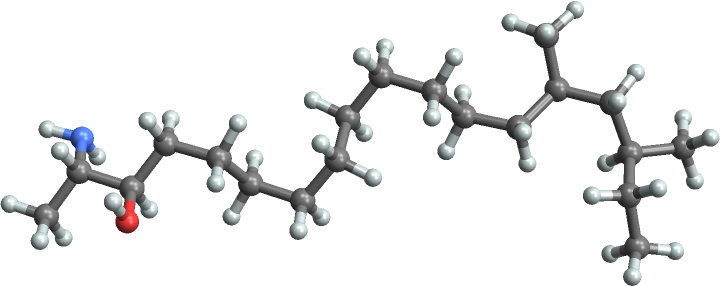 |
Use a chemical name with defined stereochemistry:
| In[3]:= |
| Out[3]= |
Plot the molecule in 2D:
| In[4]:= |
| Out[4]= | 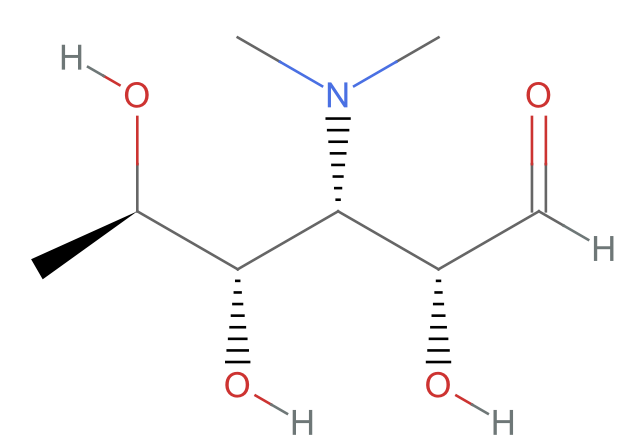 |
When given an invalid chemical name, ChemicalNameToSMILES will return a Failure identifying the uninterpretable portion of the name:
| In[5]:= |
| Out[5]= |  |
Fixing the typographical error allows the name to be parsed:
| In[6]:= |
| Out[6]= |
When parsing a chemical substituent, it is possible to add dummy atoms at the relevant attachment points:
| In[7]:= |
| Out[7]= |
By default, ChemicalNameToSMILES will parse fragment names such as "oxo" and "keto" as radicals with unpaired electrons. Use the option "AllowRadicals" to disable this behavior:
| In[8]:= |
| Out[8]= |  |
SMILES strings are canonicalized by default, so these two equivalent names return the same result:
| In[9]:= |
| Out[9]= |
Use the "Canonicalize" option to disable this:
| In[10]:= |
| Out[10]= |
This work is licensed under a Creative Commons Attribution 4.0 International License